Two systems for counting nucleotides in a cell is not impossible. While no evidence exists in the micromachinery of transcription, we find nothing contradictory. To retrofit ternary with binary logic, a protein interacting with DNA or RNA would require a first and second nucleotide encounter to determine a third. Then a lazy count of any 3 nucleotides could also render a more robust mRNA codon for the tRNA ternary of translation.
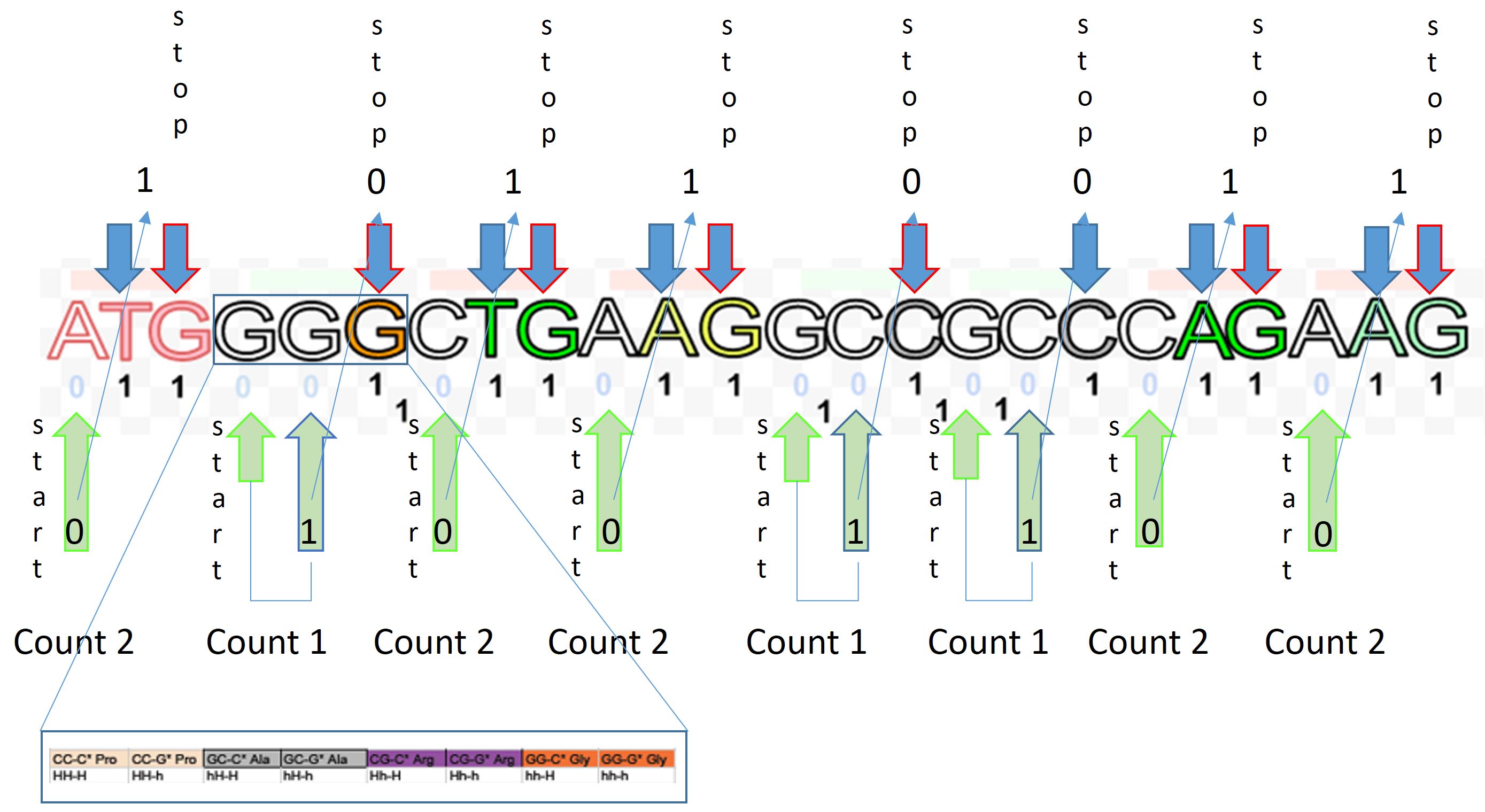
The illustration suggests an A or T encounter would ‘excite’ protein reading the next two nucleotides. However, a G or C would ‘relax’ protein, becoming excited at the next A or T encounter or at the third G or C nucleotide, but not a G or C at the second. This retrofit pretense, even if counted differently during translation reconciles with standard dogma.
If reflected in biophysics, it would challenge bioinformatics. Nevertheless, we leverage this opportunity to explore the possibility that big data procedures are presently inadequate. To model and learn every potential state, sequences have to be amplified into constituent subsequences that may have been present in a cell and analyzed to project ‘near reality’ of the cell state onto subsequences.
In one example, a Tp53 ncDNA sequence of 400 nucleotides produced more than 77,000 different, theoretical subsequences of varying lengths up to 400. We imagine any of these could be present in single cells at some time. Then, we count each subsequences’ recurrence in every other subsequence to determine its potential statistical state.
To project ‘near reality’ cell state for ncDNA subsequences, we associate the signature of the transcript cDNA/mRNA or Protein with each. Then, for each of multiple transcripts, a vector records the order each subsequence confers to the transcript using its recurrence and transcript protein signature. From this we report vector order changes by subsequences for each ‘subsequence-protein signature’ pair in the set of all transcripts.
What we do differently?
For a given sequence of a transcript we iterate single nucleotides, tag every adjacent subsequence (of length >7) and count their recurrence. We report:
- Codondex i-Score, the recurrence of a subsequence in the set of all subsequences, scored after adjusting for variable lengths >7.
- Reverse complements* and inverted repeats including subsequence start-end positions in the origin sequence
- PhV and PiV as a measure of transcripts subsequence relativity where intron(subsequence)-protein (or mRNA) pairs are described using a vector
- miRNA blasts from mirbase.org for each subsequence
- Same two letter starts compares all subsequences beginning with the same two letters
- Identical subsequences in any transcripts of any gene analyzed are identified
Why do we do this?
Ultimately we envision faster compute times using subsequence recurrence to produce your desired feature sets.
Codondex i-Score maps any subsequence recurrence (Inclusiveness)
Intron-protein pairs distinguish relative phenotypic characterizations for each iterated, noncoding subsequence
We want to process your sequences or multi-transcript intron and mRNA/protein data through our pattern algorithm that amplifies, splits and permanently pairs each subsequence with the transcript mRNA or protein. This ensures the mRNA or protein is the biological constant for the set of subsequences.
How do we validate the data?
Our digital descriptions based on recurrence of each subsequence are used to precisely and rapidly predict biological confirmations.
Example: We counted the recurrence of reverse complement text in subsequence text of transcripts. Then, using only recurrence data (derived exclusively from subsequence text) we accurately predicted the frequency of each reverse complement-inverted repeat in each subsequence. We ran prediction errors through Random Forest and resolve at or near 100% precision for each transcript.